Basic Usage#
Raw And Load Usage#
Experiment-scoped commands (everything that takes a single experiment config) live under
smftools experiment. Project-scoped commands (everything that spans multiple registered
experiments) live under smftools project. Anything outside those two groups (e.g.
subsample-pod5) is a standalone utility with no experiment/project config of its own.
In v2, workflows begin by preparing BAM artifacts and the ragged per-read source of truth:
smftools experiment raw "/Path_to_experiment_config.csv"
Dense reference-grid data is materialized on demand. To persist the full dense zarr cache ahead of downstream work, run the optional command:
smftools experiment load "/Path_to_experiment_config.csv"
This command takes a user passed config file handling: - I/O pathes (With data input path, FASTA path, optional BED path for subsampling FASTA, and a data output path) - Experiment info (SMF modality, sequencer type, barcoding kit if nanopore, sample sheet with metadata mapping) - Options to override default workflow parameters from smftools/config. Params are handled from default.yaml -> modality_type.yaml -> user passed config.csv.
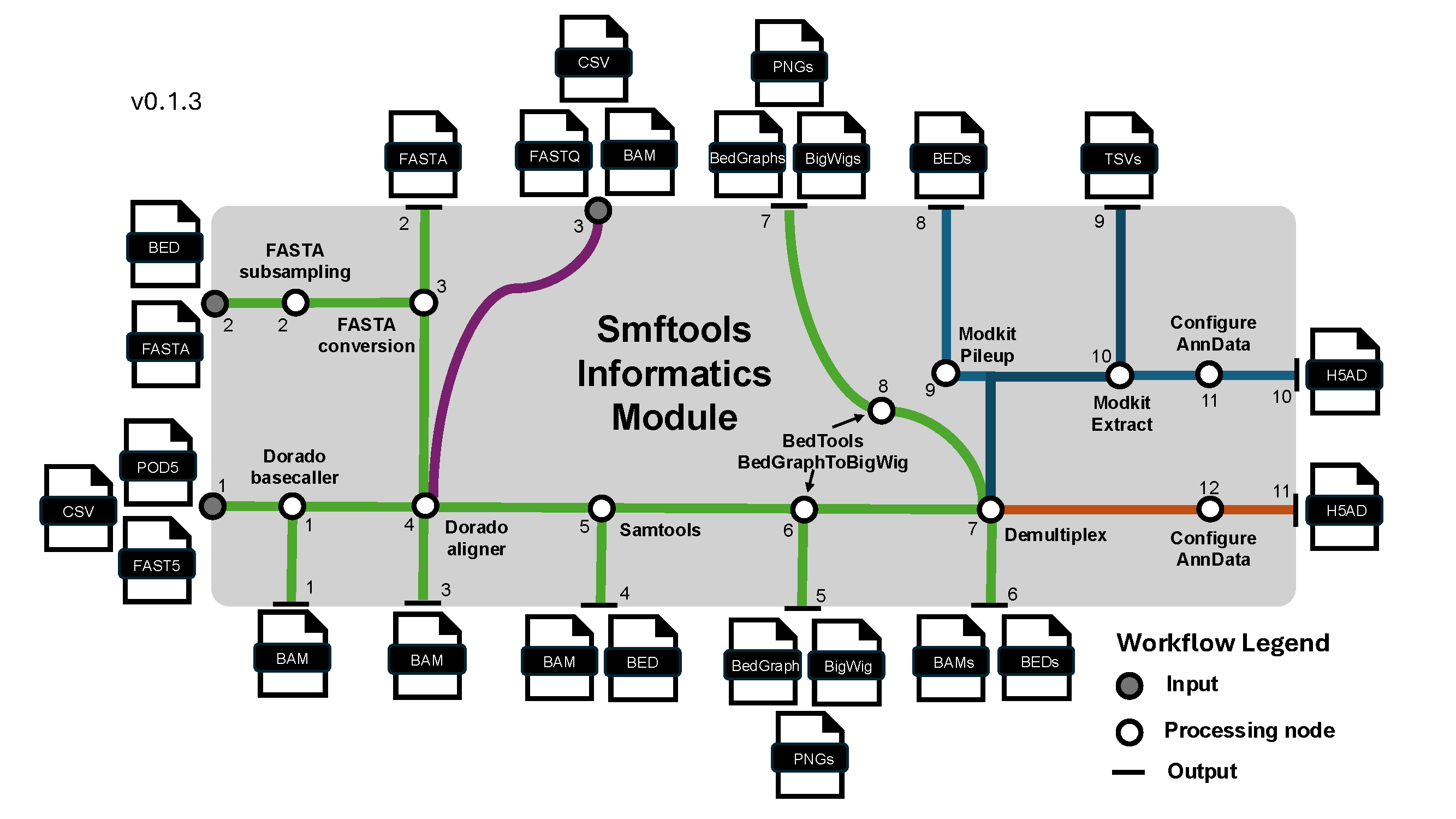
For smf_modality: deaminase experiments, raw also derives per-read strand-switch metrics
(ct_event_count, ga_event_count, strand_segment_purity) from the alignment CIGAR and, unless
bypass_raw_chimera_rate_plot is set, writes a reference x barcode heatmap of the resulting
PCR-chimera rate to raw_outputs/plots/reference_barcode_chimera_rate.png (with a companion
.csv table) so you can spot problem barcodes before preprocessing.
Preprocess Usage#
This command performs preprocessing on the anndata object.
smftools experiment preprocess "/Path_to_experiment_config.csv"
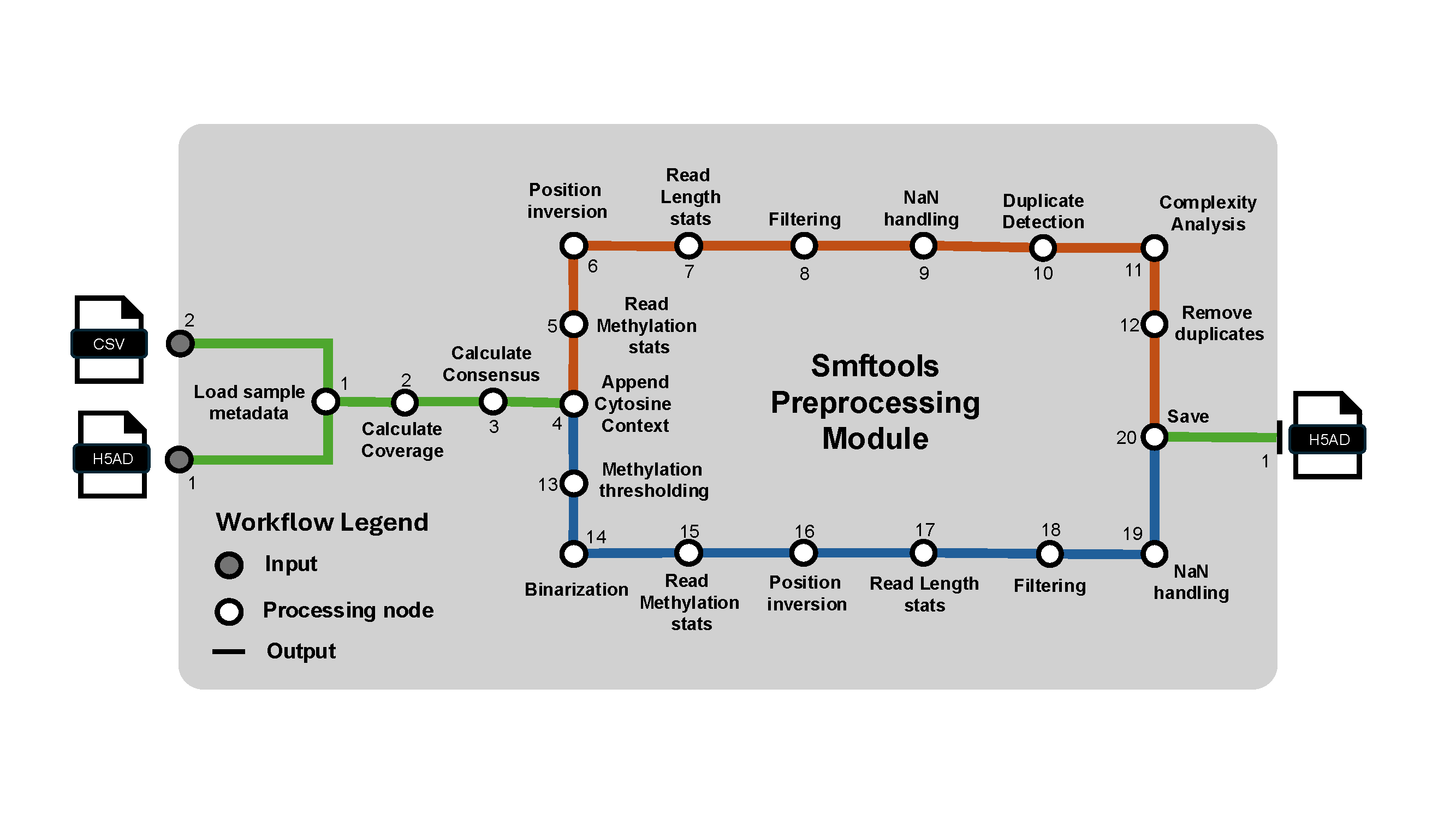
Two read-level filters/labels run as part of this stage:
CIGAR-based internal indel filtering. Reads whose longest internal insertion or deletion (from the alignment CIGAR) exceeds
max_internal_insertion_length/max_internal_deletion_length(default 10bp each) are dropped. Set either tonullto disable that check, or setbypass_filter_reads_on_cigar_indels: Trueto skip the step entirely.Deaminase PCR-chimera labeling (
smf_modality: deaminaseonly). Reads whose deamination signature switches from a C->T span to a G->A span partway through the read (evidence of two templates fused during PCR) are flagged inobs["deaminase_PCR_chimera"]-- these reads are labeled, not removed, so you can filter or inspect them downstream. Sensitivity is controlled bydeaminase_chimera_min_events_per_span,deaminase_chimera_min_segment_purity, anddeaminase_chimera_max_single_strand_fraction; setbypass_label_deaminase_pcr_chimeras: Trueto skip.
Variant Usage#
This command performs DNA sequence variation based analyses on the anndata object.
smftools experiment variant "/Path_to_experiment_config.csv"
Chimeric Usage#
This command performs putative PCR chimera detection on the anndata object.
smftools experiment chimeric "/Path_to_experiment_config.csv"
Spatial Usage#
This command performs spatial analysis on preprocessed data.
smftools experiment spatial "/Path_to_experiment_config.csv"
When a partitioned preprocessing spine is available, spatial analysis automatically runs as
bounded reference/window/barcode tasks over QC-passing deduplicated reads. It writes a linked
spatial spine, barcode-stratified autocorrelation tables, position profiles, and registered plots.
Set spatial_execution_mode to auto, partitioned, or legacy to control selection.
Locus-mode references automatically receive full-reference clustermaps and position matrices per
reference strand. For genome-mode references, set spatial_regions_bed to a standard 0-based,
half-open BED file; dense products are then limited to those intervals. BED chromosome names may
be exact reference-strand names or native reference names that apply to both strands.
Partitioned spatial analysis also saves read-level ACF and direct-signal Lomb-Scargle results by
default. Each task directory in the general spatial store contains a read_metrics.zarr AnnData
partition. Per-read ACF, pair-count, and normalized periodogram arrays use obsm; peak period,
peak power, raw peak power, SNR, FWHM, site count, and scoring status use obs; lag and
frequency/period coordinates use uns. This matches preprocessing-store conventions without
expanding the thin spatial spine. Automated periodicity plots summarize peak-period and peak-power
distributions, scoring fraction, and barcode-mean spectra for each reference region and site type.
Per-reference-and-barcode read clustermaps show the individual ACF and Lomb-Scargle power profiles
with aligned mean curves above each heatmap.
The legacy dense path continues to provide whole-reference position correlation matrices, clustermaps, and the existing AnnData-embedded spatial results.
HMM Usage#
This command performs hmm based feature annotation on the anndata object.
smftools experiment hmm "/Path_to_experiment_config.csv"
hmm_execution_mode accepts auto, partitioned, or legacy. The default auto mode uses a
partitioned spatial/preprocessing spine when available and writes bounded HMM task Zarr groups, a
task catalog, model store, plot catalog, and linked thin spine under hmm_adata_outputs.
Main outputs wills be stored in adata.layers
Latent Usage#
This command constructs various latent representations of the anndata object.
smftools experiment latent "/Path_to_experiment_config.csv"
Full Usage#
This command runs the standard workflow in order: raw store creation, preprocessing, spatial analysis, and HMM annotation. Each stage retains its normal restart/skip behavior, so an existing valid stage output is reused unless its force-redo configuration is enabled.
smftools experiment full "/Path_to_experiment_config.csv"
Batch Usage#
This command performs batch processing of any of the above commands across multiple experiments. It takes in a tsv, txt, or csv of experiment specific config csvs.
smftools experiment batch preprocess "/Path_to_experiment_config_path_list.csv"
Nice when analyzing multiple experiments
Project Usage#
A project is a lightweight cross-experiment registry: it never copies or merges data, it just
keeps pointers to experiment output directories (each containing a spine.h5ad) plus a table that
harmonizes reference names across experiments (by sequence identity, so the same locus can be
called different things in different experiments). Use it when you want to query or analyze
multiple experiments together without materializing one giant combined AnnData ahead of time. See
Organizing data, experiments, and projects for a full
directory layout, a step-by-step experiment-to-project walkthrough, and how to move or share that
directory tree across machines.
# Create a project registry.
smftools project init "/Path_to_project_directory"
# Register an experiment by pointer (its output directory -- or one stage dir
# inside it, e.g. raw_outputs -- either works, every stage found is recorded).
smftools project add "/Path_to_project_directory" "/Path_to_experiment_output_dir"
# List registered experiments (with which stages each has reached) and harmonized references.
smftools project list "/Path_to_project_directory"
# Materialize one canonical reference across every matching experiment into a single AnnData.
smftools project materialize "/Path_to_project_directory" my_canonical_reference \
-o "/Path_to_output.h5ad.gz"
# Mark an experiment inactive (soft delete; registry entries are append-only).
smftools project remove "/Path_to_project_directory" experiment_id
project adddiscovers every pipeline stage spine under the given directory and reads the raw spine'sunsmetadata (modality, sequence-hash reference identities) to register it -- no matrices are opened.project materializeresolves the canonical reference name back to each matching experiment's own reference name(s), materializes each experiment's slice independently, and concatenates them with an addedobs["experiment"]column -- there is never a global merge across experiments. Each experiment's spine is picked independently, defaulting to the most-derived stage available (--stagepins all experiments to one specific stage instead);--read-metricsadditionally attaches spatial's per-read outputs where available.smftools project export-fastq ...(below) and other cross-experiment tooling build on the same registry.
Export FASTQ Usage#
This command writes one FASTQ (gzip-compressed by default) per barcode, containing only reads that passed QC, for a single experiment or an entire project. Sequence and quality are read directly from the raw ragged store, so no BAM re-parsing is needed. It's available under both hierarchies:
# Single experiment: QC-passed read set is resolved from the most complete preprocessing
# artifact available (partitioned preprocess spine, falling back to the legacy deduplicated
# or QC-filtered AnnData).
smftools experiment export-fastq "/Path_to_experiment_config.csv" --outdir "/Path_to_fastq_output_dir"
# Whole project: writes one FASTQ per <experiment_id>__<barcode>, skipping experiments that
# have not run partitioned preprocessing.
smftools project export-fastq "/Path_to_project_directory" --outdir "/Path_to_fastq_output_dir"
--group-byoverrides the obs column used to group reads (experiment command only; defaults tosample_name_col_for_plotting, falling back toSamplethenBarcode).--experimentsrestricts the project command to a comma-separated list of experiment ids (default: all active).--allow-unfilteredwrites every raw read instead of raising/skipping when no QC-passed read set is available yet (i.e. beforesmftools experiment preprocesshas been run).--no-gzipwrites plain.fastqinstead of.fastq.gz.A
fastq_manifest.csv(barcode, read count, output path) is written alongside the FASTQs.
Concatenate Usage#
This command concatenates multiple h5ad files and saves them to a new output. The h5ads to concatenate are provided as a txt, tsv, or h5ad file of paths.
smftools experiment concatenate "/Path_to_experiment_config.csv" -c "/Path_to_h5ad_path_list.csv"
Alternatively, you can just concatenate all h5ads within a given directory.
smftools experiment concatenate "/Path_to_experiment_config.csv" -d "/Path_to_h5ad_file_dir/"
Mainly used for combining multiple experiments into a single anndata object.
Subsample POD5 Usage#
This command subsamples a POD5 file or a directory of POD5 files. It can be done by passing a txt file of read names to use, or an integer number of reads.
smftools subsample-pod5 -r "/Path_to_read_name_list.txt" -o "/Path_to_output_directory" "/Path_to_input_POD5_dir_or_file"
smftools subsample-pod5 -n 1000 -o "/Path_to_output_directory" "/Path_to_input_POD5_dir_or_file"
Optional run logging#
If you want to maintain run log files of CLI processes, you can use the following syntax to any of the CLI commands. Here is an example using smftools experiment load with logging performed on INFO level logging outputs and above.
smftools --log-file "/Path_to_output_log_file.log" --log-level INFO experiment load "/Path_to_input_config.csv"
Reading AnnData objects created by smftools#
After creating an AnnData object holding your experiment's SMF data, you can load the AnnData object as so:
import smftools as smf
input_adata = "/Path_to_experiment_AnnData.h5ad.gz"
adata = safe_read_h5ad(input_adata)
This custom read function will take an optional directory of pickle files for data types that can not normally be saved directly in hdf5 formatting that was saved with the safe_write_h5ad function.
If you don't have an AnnData object yet, but want to play with the downstream Preprocessing, Tools, and Plotting modules, you can load a pre-loaded SMF dataset.
Currently, you can do this with our lab's in vitro dCas9 binding kinetics dataset generated from a Hia5 SMF dataset generated with direct m6A high accuracy basecalls:
adata = smf.datasets.dCas9_kinetics()
adata.obs_names_make_unique()
Alternatively, you can do this with our lab's M.CviPI SMF test data in F1-hybrid natural killer cells generated by NEB EMseq conversion followed by canonical basecalling:
adata = smf.datasets.Kissiov_and_McKenna_2025()
adata.obs_names_make_unique()
Writing out AnnData objects to save analysis progress#
After preprocessing and downstream analysis of the AnnData object, you can save the AnnData object at any step as so:
import smftools as smf
from pathlib import Path
output_dir = Path('/Path_to_output_directory')
output_adata = 'analyzed_adata.h5ad.gz'
final_output_path = output_dir / output_adata
safe_write_h5ad(adata, final_output_path, compression='gzip')
This custom save function will make a directory of pickle files for data types that can not normally be saved directly in hdf5 formatting.
Troubleshooting#
For more advanced usage and help troubleshooting, the API and tutorials for each of the modules is still being developed. However, you can currently learn about the functions contained within the module by calling:
smf.inform.__all__
This lists the functions within any given module. If you want to see the associated docstring for a given function, here is an example:
print(smf.inform.load_adata.__doc__)
These docstrings will provide a brief description of the function and also tell you the input parameters and what the function returns. In some cases, usage examples will also be provided in the docstring in the form of doctests.